致细胞深处的"无名英雄":守护我们一生的线粒体"三人组"
发布时间:2026-05-11
你有没有想过,为什么我们的亲人会在年老时罹患帕金森氏症?为什么有些朋友年纪轻轻就行动迟缓、说话不清?为什么有些孩子一出生就面临多重发育障碍?答案,可能藏在我们身体细胞里的"发电站"中。
我们的大脑和我们的身体一样,都需要"发电站"提供能量。如果把我们的大脑比作小宇宙,那么组成它的860亿个神经元细胞,每一个都是一座精密运转的城市,而在每一座神经元城市里,都建有成百上千座被称为"线粒体"的发电站,它们彼此连接成网,24小时为细胞生产能量,让我们能够自由奔跑、畅想、感受这个世界。但是你知道吗?每座发电站内部还藏着一支由"AAA+蛋白酶"组成的精锐维修部队,它们分别驻扎在不同的车间,负责清理故障零件、修复受损设备,日夜守护着能量生产的稳定运转。一旦这支队伍失职,帕金森病、阿尔茨海默病、脊髓小脑共济失调,甚至严重的神经发育疾病……就会悄然逼近[1]。今天,我们就来认识一下这支维修部队中的三位"守护者"。
发电站没人打扫会怎样
家里的垃圾如果一周不清理,会怎样?臭气熏天,滋生细菌,甚至引来蟑螂!细胞里的发电站也一样。线粒体蛋白质组时刻面临各种应激反应:高温、氧化损伤、合成错误,都会催生大量异常折叠的蛋白质。如果这些错误折叠的"垃圾蛋白质"不及时处理,就会干扰发电设备,也就是线粒体DNA的复制/转录和线粒体呼吸链复合物的组装,甚至还可能触发多种细胞死亡程序[1]。这就像发电站里堆满了废料,机器无法运转,最终整个电站不得不停工。这就是为什么线粒体需要一支专业的"清道夫队伍"--线粒体AAA+蛋白酶。
三位"守护者"的身份档案
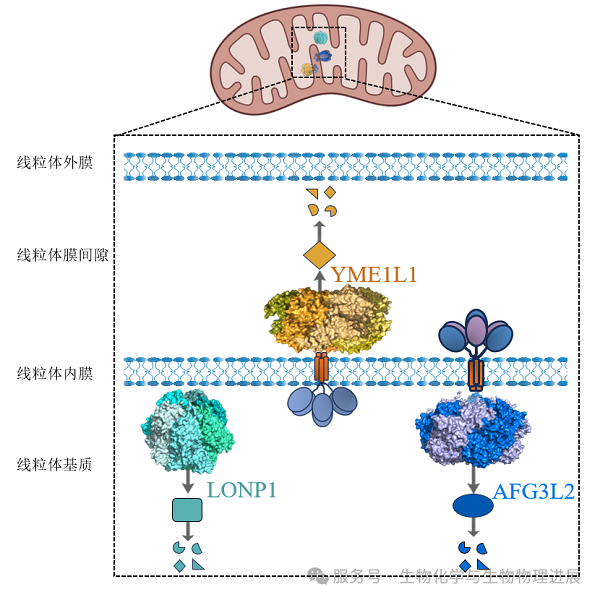
在人类细胞的线粒体中,目前已鉴定出超过1158种蛋白质,它们精准定位于线粒体的四个功能区室[1]。不同的AAA+蛋白酶分布于不同区域,形成功能互补的质控网络。其中,有三类蛋白酶构成了质控网络的核心。让我们来认识一下这三位默默工作的"守护者"。
基质的"全能清洁工"--Lon蛋白酶1(Lon peptidase 1,LONP1)[2]。LONP1主要定位于线粒体基质(发电站核心区),其核心功能主要包括:降解错误折叠蛋白、维持线粒体DNA的稳定性、应激时优化呼吸链的反应效率。
膜间隙的"质检员"--酵母线粒体逃逸基因1样蛋白1(yeast mitochondrial escape 1 like 1,YME1L1)[3]。YME1L1锚定于线粒体内膜,面向内膜和外膜之间的膜间隙,其核心功能主要为:调控线粒体融合、监控蛋白质输入通道、降解带有"报废标签"的底物。
内膜的"双面维修工"--ATP酶家族基因3样蛋白2(ATPase family gene 3-like 2,AFG3L2)[4]。AFG3L2锚定于线粒体内膜,面向基质,主要参与呼吸链复合物的组装、维持线粒体核糖体稳态、调控钙信号等过程。
当"守护者"失职:从分子缺陷到神经系统疾病
神经系统对能量供应高度依赖,神经元中超过90%的ATP由线粒体提供。因此,当这些蛋白酶"守护者"出现问题时,最先倒下的往往是我们的大脑[5]。值得注意的是,"守护者"失职的严重程度决定了疾病的发病时间:个别情况下从出生时就可能出现异常,但所有个体到达老年期一定会逐渐衰退,因此线粒体质量控制系统保护细胞生命的全过程。
01 出生时即崩溃:CODAS综合征--发电站还没建成就"断电"
有些婴儿一出生就面临多重挑战:头颅和面部的骨骼发育异常、牙齿长不出来、耳朵和眼睛的结构缺陷,甚至连四肢的骨骼都在慢慢变形。这不是普通的先天缺陷,而是一种极其罕见的疾病,称为脑、眼、牙、耳、骨骼异常(cerebral, ocular, dental, auricular,skeletal anomalies,CODAS)综合征[6]。它的病因很清晰:在细胞发电站建造的关键时期,负责清理建筑垃圾的"全能清洁工"LONP1完全罢工。
正常情况下,LONP1在线粒体基质中负责降解错误折叠蛋白,维持发电站有序运转。但CODAS患儿的LONP1基因发生了突变,使LONP1的六聚体结构变得松散,ATP酶活性显著下降。这导致线粒体能量生产严重不足,因此使大脑、眼睛、骨骼等能量需求高的器官发育失败[7]。这就是CODAS综合征的真相:从生命之初,能量系统就崩溃了。
02 神经发育期的困境:YME1L1与孤独症--当"质检员"出错,社交的桥梁断裂
孤独症谱系障碍(autism spectrum disorder,ASD)是一种神经发育疾病,核心表现为社交障碍、重复刻板行为和兴趣狭窄。它的背后,可能藏着线粒体"质检员"YME1L1的失职。
在一项针对116个ASD家系的大规模基因组学研究中,科学家们系统分析了罕见基因变异与ASD的关联,识别出多个新的潜在风险基因,其中就包括YME1L1[8]。研究聚焦于一个编号117的家系。家系中的先证者是一名早发性孤独症患儿,伴有语言发育异常、轻度智力障碍和局灶性癫痫。基因测序发现,他携带一个YME1L1的新生错义突变(即新发生的、导致氨基酸改变的基因突变)--c.1981G>A,导致蛋白质第661位的谷氨酸被赖氨酸替换[8]。这一发现提示:当YME1L1这把"分子剪刀"出现故障,线粒体质量控制系统被打乱,最终可能影响神经元发育,将孩子困在自己孤独的世界里。
03 中年突遭变故:脊髓小脑共济失调28型--身体突然"不听指挥"了
你有没有见过这样的人:说话含糊不清,类似醉酒状态;拿东西时手抖得厉害;走路步子宽宽的、摇摇晃晃,像随时要摔倒。这不是喝醉了,也不是自然衰老退化,而是一种称为脊髓小脑共济失调28型(spinocerebellar ataxia 28,SCA28)的遗传病。它通常在30~50岁之间悄悄起病,慢慢剥夺一个人的运动协调能力[9]。
其问题出在AFG3L2基因上。患者只有一份正常基因,另一份突变了,导致AFG3L2蛋白产量减半。部队中的AFG3L2士兵数量锐减,只能勉强维持发电站的日常运转,但随着年龄增长,问题逐渐暴露:线粒体出现碎片化;复合物Ⅳ活性降低约40%;ATP合成减少35%。当能量持续供应不足,小脑的浦肯野细胞最先撑不住,它们开始功能失常,然后慢慢死去[9]。于是,身体渐渐"不听指挥"了:想说话,舌头不听使唤;想拿水杯,手在抖;想走直线,腿在晃。这不是老了,是发电站供不上电了。
04 老年期的慢性侵蚀:阿尔茨海默病--当记忆被"偷走"时,线粒体里藏着的真相
你是否见过这样的老人:刚刚吃过饭就忘了,反复问同一件事;走过几十年的路也会迷失;曾经开朗的性格变得多疑、暴躁。这是阿尔茨海默病,一种悄悄偷走记忆和性格的疾病[10]。
它为什么偏偏攻击大脑?研究发现,在AD患者的大脑深处,一种叫"β淀粉样蛋白(Aβ42)"的异常蛋白会潜入线粒体,与LONP1相互作用并抑制其活性,导致抗氧化酶堆积、氧化应激水平飙升、ATP产量下降40%,最终导致神经元死亡,记忆就这样被一点点抹去[11]。

05 老年期的运动崩塌:帕金森病--身体开始"不听使唤"
你有没有注意过一些老人手会不由自主地抖动,像在打拍子?走路时步子很小、身体前倾,好像随时会摔倒?这是帕金森病的典型表现:运动迟缓、静止性震颤、肌强直[12]。
问题的源头也在细胞发电站。约5%~10%的帕金森病患者有家族遗传背景,其中一种早发型PD与DJ-1基因突变有关。突变后的DJ-1蛋白发生错误折叠,如果发电站的清洁工LONP1功能异常,这些错误折叠的DJ-1蛋白聚集体就会堆积成山。结果导致:线粒体呼吸链复合物Ⅰ被抑制,能量生产受阻;活性氧类大量产生,毒害周围环境;多巴胺能神经元开始死亡[12]。而这些多巴胺能神经元负责调控我们的运动,它们一批批死去,身体就渐渐"不听使唤",先是一只手抖,然后一条腿拖,最终全身僵硬。
治疗前景:我们能唤醒沉睡的"守护者"吗
了解了这些"守护者"如何失职、如何引发疾病,一个自然的问题摆在面前:我们能修复它们吗?答案是:科学家们正在努力,但挑战依旧重重。
01 实验室里的发现
研究人员已经发现了一些可以调控LONP1活性的化合物。
CDDO及其衍生物:它们像一把"分子开关",通过阻断ATP结合抑制LONP1活性[13]。这为癌症等需要"踩刹车"的疾病提供了思路,但抑制LONP1可能加剧神经退行性风险,这把双刃剑需要谨慎使用。
青蒿素类药物(如双氢青蒿素DHA、青蒿琥酯ART):它们能调控LONP1与特定蛋白质的相互作用,为多囊卵巢综合征打开了新的治疗窗口[14]。
02 为什么调控如此困难
这些"守护者"处于线粒体质量控制网络的核心,它们的活性必须维持精密的平衡:激活过度会误伤必需蛋白质,导致正常功能受损;抑制过度则使毒物堆积,细胞仍难逃死亡命运。
03 未来的突破方向
科学家们正在从多个角度突破。看得更清:利用原位冷冻电镜,在近生理条件下"直播"蛋白酶构象变化;靶向更准:开发针对蛋白酶特异性结构的高选择性调节剂;摸清规律:研究不同细胞类型、不同发育阶段中,这些蛋白酶的表达和调控有何差异?
结语
从CODAS患儿的第一次呼吸,到帕金森病老人的最后一次握手,线粒体里的这三把"分子剪刀"始终在幕后雕刻着生命的长度与宽度。当这些"维护系统"正常运转时,我们几乎感受不到它们的存在,而当它们失职时,疾病便悄然逼近。
下一次,当你思维清晰、步伐稳健时,不妨默默感谢一下细胞发电站里那三位默默工作的"守护者"。 而科学家们正在努力,希望让"守护者"们更好地发挥作用,让每一个生命都能拥有更长久、更健康的旅程。(详情请点击阅读原文)
参考文献
[1] Quirós P M, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol, 2015, 16(6): 345-359
[2] Lee Y G, Kim H W, Nam Y, et al. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis, 2021, 10(2): 18
[3] Wai T, García-Prieto J, Baker M J, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science, 2015, 350(6265): aad0116
[4] Caporali L, Magri S, Legati A, et al. ATPase domain AFG3L2 mutations alter OPA1 processing and cause optic neuropathy. Ann Neurol, 2020, 88(1): 18-32
[5] Wilson D M 3rd, Cookson M R, van den Bosch L, et al. Hallmarks of neurodegenerative diseases. Cell, 2023, 186(4): 693-714
[6] Shebib S M, Reed M H, Shuckett E P, et al. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome-a case report. Am J Med Genet, 1991, 40(1): 88-93
[7] Strauss K A, Jinks R N, Puffenberger E G, et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Hum Genet, 2015, 96(1): 121-135
[8] Viggiano M, Ceroni F, Visconti P, et al. Genomic analysis of 116 autism families strengthens known risk genes and highlights promising candidates. NPJ Genom Med, 2024, 9(1): 21
[9] L?bbe A M, Kang J S, Hilker R, et al. A novel missense mutation in AFG3L2 associated with late onset and slow progression of spinocerebellar ataxia type 28. J Mol Neurosci, 2014, 52(4): 493-496
[10] Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet, 2021, 397(10284): 1577-1590
[11] Wang W, Ma X, Bhatta S, et al. Intraneuronal β-amyloid impaired mitochondrial proteostasis through the impact on LONP1. Proc Natl Acad Sci U S A, 2023, 120(51): e2316823120
[12] Bonifati V, Rizzu P, van Baren M J, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science, 2003, 299(5604): 256-259
[13] Lee J, Pandey A K, Venkatesh S, et al. Inhibition of mitochondrial LonP1 protease by allosteric blockade of ATP binding and hydrolysis via CDDO and its derivatives. J Biol Chem, 2022, 298(3): 101719
[14] Liu Y, Jiang J J, Du S Y, et al. Artemisinins ameliorate polycystic ovarian syndrome by mediating LONP1-CYP11A1 interaction. Science, 2024, 384(6701): eadk5382
作者简介
李茹茹:西南医科大学与中国科学院生物物理研究所联培生。研究方向:认知障碍和运动障碍等相关脑疾病的分子机制。
朱 笠:中国科学院生物物理研究所认知科学与心理健康全国重点实验室研究员。研究方向:认知障碍和运动障碍等相关脑疾病的分子机制。
(作者:李茹茹、朱笠)
(本文来源于公众号:生物化学与生物物理进展)
附件下载:
上一篇: