高通量解析处于复杂生物环境中的蛋白质机器三维结构
发布时间:2021-09-10
2021年9月8日,《The Innovation》杂志在线发表了中国科学院生物物理研究所章新政课题组的研究成果"Determining structures in a native environment using single-particle cryo-electron microscopy images"。该工作是继2018年开发的分块重构算法(Nat Commun 9, 1552),实现蛋白质叠合密度中的结构优化后,又发展的一种叠合密度中的蛋白质识别方法,将算法推广到了原位结构解析。该算法针对处于复杂生物环境中的蛋白质分子提出了新的结构解析方法,利用无倾转数据采集方式,并通过优化蛋白质探测算法、去除模型偏差以获得目标蛋白质结构,使数据收集通量得到了十倍以上的显著提升,同时分辨率也得到了提高。
目前,针对复杂生物环境中的冷冻电镜结构解析主要是基于电子断层重构(cryo-ET)。Cryo-ET需要倾转样品台采集不同角度的序列倾转数据,通量非常低,要想获得较高分辨率的重构,往往需要进行10天以上的数据采集,这对于大部分电镜平台是难以承担的。其次,由于数据的缺乏以及断层重构技术内源性的缺陷,基于断层重构的原位结构解析技术分辨率非常受限。上述这些缺点严重限制了冷冻电镜原位结构解析研究的开展。
对处于复杂生物环境中的样品,目标蛋白在冷冻电镜图像中的密度与其他周围蛋白的密度重叠,本研究基于此开发了一种蛋白质识别算法,并对探测函数的最优权重进行了推导,新函数提升了叠合密度中探测目标蛋白的效率。结合排序函数减少蛋白质识别算法引入的假阳性数据,有效减轻模型偏差的影响,实现高分辨重构(图1)。该算法已经高通量的实现以下样品的高分辨结构解析:结合在脂质体上的膜蛋白、非二十面体病毒中的糖蛋白以及亚细胞单元中的蛋白质复合物。
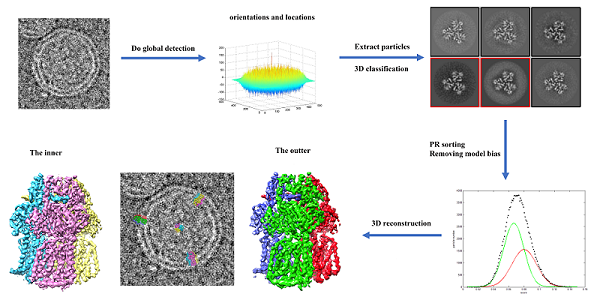
图1. 脂质体上膜蛋白的结构解析
中国科学院生物物理所章新政研究员为本文通讯作者,章新政组博士生程静为本文第一作者,博士生李不凡和中国科学院生物物理所常文瑞/李梅课题组博士生司龙也为本研究工作做出了贡献。该工作得到了国家自然科学基金委项目等的资助。
文章链接:https://www.sciencedirect.com/science/article/pii/S2666675821000916
(供稿:章新政研究组)
附件下载:
下一篇: