多肽——神经退行性疾病治疗的未来
发布时间:2022-05-06
细胞作为生物体结构和功能的基本单位,是一个“人口”密集的小型社会,其中的组分各司其职,保证生物体正常生命活动的进行。蛋白质是细胞生命活动的执行者,从微小的生化反应催化到体内免疫调控,蛋白质几乎参与细胞内所有的生物学过程。蛋白质由细胞中的“蛋白质合成工厂”——核糖体中合成的多肽链经过折叠,形成具有特定三维结构的蛋白质分子。对于一个仅含有100个氨基酸的多肽链而言,即使每个氨基酸残基只具有两种构象,多肽链可能采取的构象种类即可达到1000种以上【1】。同时,哺乳动物细胞中含有一万到两万种不同的蛋白质,细胞内蛋白质浓度可达到50~200 g/L,这也使得细胞内的环境非常拥挤【2】。在这样拥挤而忙碌的胞内环境中,蛋白质如何从多肽链高效有序地折叠成具有特定空间结构的功能态是一个令人困惑的问题。然而经过长期不断的探索,这一谜团逐渐被揭开。
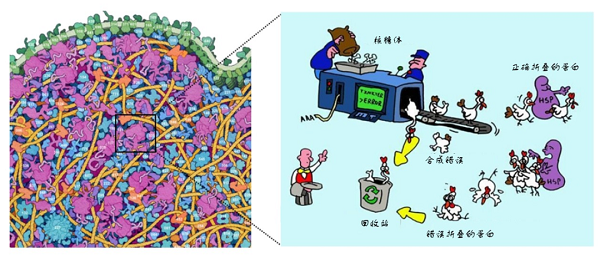
拥挤而忙碌的细胞【3】
能量景观图(energy landscape)常用来描述多肽链折叠成特定空间三维结构过程中所对应的能量变化。多肽链自身所具有的热力学和动力学特性决定了其最终构象状态,因此对于特定的氨基酸序列,它所对应的能量曲面形状是一定的。就蛋白质折叠过程而言,最初由核糖体新合成的多肽链处于高自由能和高熵值的状态,非常不稳定,而蛋白质的功能态构象往往处于热力学稳定的低自由能状态,因此肽链折叠过程就是一个自由能不断降低的过程,多肽链会沿着一个“下坡路”最终到达低自由能的功能态空间结构。但是这个“下坡路”并非一路坦途,而是崎岖和充满陷阱的。在找到正确的构象之前,多肽往往会“陷入”许多不同的中间构象,形成折叠中间体。一部分中间体处于正确路径中,可以通过调整构象变化,进而跨越出“误区”,重新进入“正途”,最终折叠形成正确的结构;但另外一部分中间体则陷入高自由能的能量陷阱发生错误折叠,甚至形成错误折叠聚集体。当聚集体大量产生而无法被细胞内蛋白质质量控制系统及时清除时,就会影响细胞的正常代谢过程。某些蛋白质由于发生错误折叠,形成高度有序的富含特征性-cross-β结构的聚集体,被称为淀粉样纤维,这种结构使蛋白质分子总体处于热力学上更为稳定的状态。蛋白质的淀粉样纤维化与常见的阿尔茨海默病(Alzheimer's disease,AD)、帕金森病(Parkinson's disease,PD)、肌萎缩性脊髓侧索硬化症(Myotrophic Lateral Sclerosis,ALS)等多种神经退行性疾病多种疾病密切相关[4]。
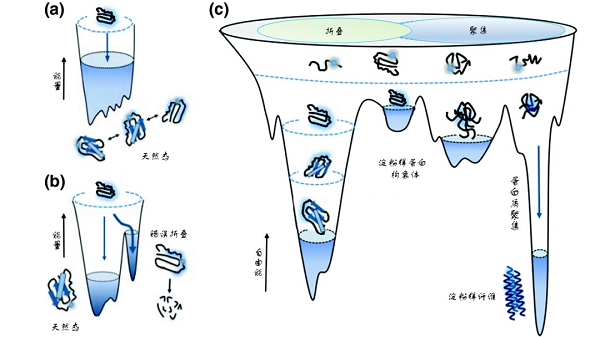
蛋白质折叠和错误折叠的能量景观图【4】
蛋白质错误折叠与神经退行性疾病
随着全球老龄化问题的日益严峻,与衰老密切相关的疾病已成为主要的公共健康危机,阿尔茨海默病(AD)、帕金森病(PD)都是以进行性认知障碍和记忆减退为主要特征的中枢神经退行性疾病。据国际阿尔茨海默病协会发布的《2021年世界阿尔茨海默病报告》显示,每3秒钟,全球就会有一位痴呆症患者产生。目前全世界约有5500多万痴呆患者,预计到本世纪中叶这一数字可能会增加到1.52亿以上,其中约70%为AD患者。目前该疾病每年造成的损失约为1万亿美元,预计到2030年这一数字将翻番【5】。PD是仅次于AD的全球第二大神经退行性疾病,PD在50岁之前比较少发,但是60~90岁之间PD的发病率会增加5~10倍。随着老龄化问题的不断加剧,预计到2050年,60岁以上人口将成为中国人口的主要组成部分,这就使得PD的发病形式变得极为严峻。目前普遍观点认为,大脑中存在的相关病理蛋白如:β淀粉样蛋白(Aβ)、微管结合蛋白Tau,以及α-突触核蛋白(α-synuclein)等的异常淀粉样纤维化与AD、PD等退行性疾病的发生发展密切相关,因此探究蛋白质发生错误折叠和淀粉样纤维化的分子机制、揭示淀粉样纤维化引发细胞毒性和病理状态的原因、开发抑制蛋白质错误折叠与聚集的有效药物,对于神经退行性疾病的防治至关重要【6】。
蛋白质错误折叠疾病的多肽药物
由于淀粉样蛋白构象的无序性、纤维化过程的动态性和复杂性,以及致病组分的不确定性,使得药物研发工作充满困难和挑战。在过去近30年间,研究者们已针对蛋白质错误折叠相关疾病发展出多种不同的治疗策略,例如:通过干预蛋白质生成相关酶的活性,降低错误折叠蛋白质的生成水平;通过靶向识别错误折叠蛋白质或聚集体,将对错误折叠蛋白进行特异性降解;增强和激活蛋白质质量控制体系以减少蛋白质错误折叠和淀粉样纤维化;清除已形成的淀粉样纤维聚集体、阻断错误折叠聚集体在细胞间传播等。相关的药物主要包括抗体、有机小分子和多肽等三大类【7】。其中多肽药物是由多个氨基酸通过肽键连接而形成的一类分子,通常由10~100个氨基酸组成,相对分子质量低于10 ku,因其高效性、可变和易修饰性、可降解性等诸多优势而备受关注。
相比于小分子药物,多肽主要通过水解和肾过滤来清除,水解的产物为氨基酸,因此多肽药物的代谢产物毒性很低;其次多肽类药物往往以内源性多肽为模板进行特异性设计,通常具有较高的靶标亲和力。而与大分子蛋白药物相比,多肽化学合成技术成熟,容易与杂质或副产物分离,并且容易引入非天然氨基酸,在质量、纯度、产量、成本等方面均具有优势。对比体积较大的抗体药物,多肽则具有较好的膜透过性、血脑屏障通过率以及较低的免疫原性,因而有望成为治疗蛋白质错误折叠和聚集相关疾病的候选药物【8】。
多肽类药物可分为天然多肽序列和多肽修饰物或类似物,主要以抑制淀粉样纤维化发生、减慢纤维聚集体生长、降低淀粉样纤维及寡聚体的细胞毒性、促进高毒性寡聚体转化为低毒性组分为目的。其中天然多肽通过对淀粉样蛋白成纤维核心的关键多肽序列进行筛选,从而获得有效的抑制剂前体,是一种常见的多肽抑制剂设计策略。最早在Aβ淀粉样纤维化的研究中发现,Aβ纤维核心序列——Aβ五肽片段KLVFF,能够与全长Aβ40结合,破坏Aβ单体之间的有序组装,提示成纤维核心序列肽可对Aβ的纤维化产生抑制作用。以此为基础对KLVFF多肽进行改造,在保持对成纤维核心序列识别能力的同时,增强多肽抑制剂的溶解性,加入破坏链间β折叠的氨基酸及其他修饰,引起聚集形式改变,降低Aβ的细胞毒性【9】。而多肽类似物则是研究者鉴于成纤维核心序列本身,往往具有较强的疏水性和聚集倾向,为提高多肽抑制剂的稳定性、水溶性和抗水解性,研究者对其进行了修饰和改造,主要手段包括:氨基甲基化修饰、D型多肽、多肽的环化等。其中链骨架上的氨基(—NH—)甲基化能够阻断链间氢键网络的形成,因而有效抑制淀粉样纤维化;同时,氨基甲基化多肽具有更高的水溶性和磷脂膜穿透能力【10】。天然氨基酸均为L型,但是L型多肽序列很容易被生物体内各种蛋白酶降解,使得多肽效率降低。而D型氨基酸构成的多肽序列则能够有效抵抗体内蛋白酶的水解作用,进而提高多肽抑制剂的代谢稳定性,同时降低免疫原性,增强通过血脑屏障的能力。已有的研究发现,抑制Aβ纤维化的多肽KLVFFA的D型对映异构体klvffa,比天然L型多肽具有更高的抑制纤维化能力,在Tau和α-syn多肽抑制剂研究中,基于纤维核心结构设计特异性结合的D型多肽也被作为一种重要策略【11】。而多肽环化则是通过限制多肽的构象,提高肽抑制剂的稳定性。研究人员通过将Aβ(1~28)中的Lys17和Asp21的侧链形成酰胺键,合成含有环结构的多肽cyclo(17,21)-Aβ(1~28),环化后的多肽自身不会聚集,且可抑制Aβ的聚集,进而降低其细胞毒性【12】。除上述策略外,还可对多肽抑制剂进行其他化学修饰,或将其连接到纳米颗粒载体表面,以增强多肽抑制剂的稳定性和抗水解性,提高其血脑屏障通过率,以达到更好的抑制效果。此外,将具有识别和抑制效应的多肽序列插入抗体中,可特异性结合淀粉样蛋白单体及其聚集物,为抗体药物的开发提供新思路【13】。
展望与前景
多肽是较早开始发展的针对蛋白质错误折叠相关疾病的药物前体,截至目前已开展了大量的研究工作。因多肽具有高特异性、低毒性、低免疫原性、易修饰性,以及生物兼容性等优势,它们有望成为治疗退行性疾病的先导分子和备选药物。然而,目前多数研究仅停留在体外实验及细胞实验水平,仅有少数多肽进入临床前研究,尚未成为临床确证有效的药物。尽管退行性疾病的致病机制尚未完全清楚,仍然存在争论,但大量的研究证据表明,蛋白质淀粉样聚集的过程与退行性疾病的发病密切相关【14】。鉴定蛋白质聚集过程中不同组分的细胞毒性及其时空因果关系,深入揭示蛋白质聚集体的结构及其与毒性的关联,对于发展有效的治疗策略起关键作用。在未来的研究中,包括干扰淀粉样纤维化过程在内的多种治疗策略的联合应用将有望为退行性疾病治疗带来新希望。同时,基于淀粉样纤维化过程开发疾病检测和诊断的生物标志物,对于退行性疾病的早期诊断以及评判临床治疗效果也有十分重要的意义。(详情请点击阅读原文)
参考文献
[1] Anfinsen C B. Principles that govern the folding of protein chains. Science (New York, NY), 1973, 181(4096): 223-230
[2] Phillip Y, Schreiber G. Formation of protein complexes in crowded environments – from in vitro to in vivo. FEBS Lett, 2013, 587(8): 1046-1052
[3] Goodsell D S, Olson A J, Forli S. Art and science of the cellular mesoscale. Trends Biochem Sci, 2020, 45(6): 472-483
[4] Parry T L, Melehani J H, Ranek M J, et al. Functional amyloid signaling via the inflammasome, necrosome, and signalosome: new therapeutic targets in heart failure. Front Cardiovasc Med, 2015, 2:25
[5] Https://www.Alzint.Org/Resource/World-Alzheimer-Report-2021/
[6] Balestrino R, Schapira A H V. Parkinson disease. Eur J Neurol, 2020, 27(1): 27-42
[7] Naddaf E, Barohn R J, Dimachkie M M. Inclusion body myositis: update on pathogenesis and treatment. Neurotherapeutics, 2018, 15(4): 995-1005
[8] Armiento V, Spanopoulou A, Kapurniotu A. Peptide-based molecular strategies to interfere with protein misfolding, aggregation, and cell degeneration. Angew Chem Int Ed Engl, 2020, 59(9): 3372-3384
[9] Tjernberg L O, N?slund J, Lindqvist F, et al. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem, 1996, 271(15): 8545-8548
[10] Gordon D J, Tappe R, Meredith S C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1-40 fibrillogenesis. J Pept Res, 2002, 60(1): 37-55
[11] Sievers S A, Karanicolas J, Chang H W, et al. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature, 2011, 475(7354): 96-100
[12] Luo Q, Lin Y X, Yang P P, et al. A self-destructive nanosweeper that captures and clears amyloid β-peptides. Nat Commun, 2018, 9(1): 1802
[13] Sormanni P, Aprile F A, Vendruscolo M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc Natl Acad Sci USA, 2015, 112(32): 9902-9907
[14] Eisele Y S, Monteiro C, Fearns C, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov, 2015, 14(11): 759-780
作者简介
温纪涛:中国科学院生物物理研究所博士后,目前从事淀粉样蛋白聚集分子机制研究。
(作者:温纪涛)
(本文来源于公众号: 生物化学与生物物理进展)
附件下载:
上一篇:
下一篇: