朱笠研究组科研进展:线粒体自噬受体FUNDC1通过介导线粒体
蛋白输入和线粒体质量控制参与神经退行性疾病中TDP-43的降解
发布时间:2023-11-19
TDP-43蛋白病是以TDP-43蛋白的异常沉积为神经病理学特征的一系列神经退行性疾病的总称,包括肌萎缩侧索硬化症(ALS-TDP)、额颞叶变性病(FTLD-TDP)、阿尔茨海默病(AD-TDP)和边缘主导型年龄相关 TDP-43脑病(LATE-TDP)等。TDP-43蛋白的表达异常与蛋白质稳态调控失衡可能是TDP-43蛋白病的发病机制。中国科学院生物物理研究所的研究团队一直关注并研究TDP-43参与神经退行性疾病的分子机制。他们发现TDP-43疾病相关突变多肽不仅自身形成淀粉样纤维,而且能诱导或加速不能形成纤维的其他TDP-43多肽及AD相关多肽A 1-40发生纤维化;运用NMR方法解析TDP-43突变多肽的结构,发现其呈反平行 结构,为其在体外形成淀粉样纤维和在神经元细胞内诱导TDP-43蛋白从细胞核重新分布至细胞质并造成神经元死亡提供了结构基础(Zhu et al., Human Molecular Genetics,2014)。利用FTLD-TDP患者脑样本结合TDP-43蛋白病的细胞模型和转基因果蝇模型进行研究,发现TDP-43可进入线粒体并激活线粒体去折叠蛋白反应(UPRmt),UPRmt相关的线粒体蛋白酶LONP1参与降解TDP-43蛋白,下调LONP1的表达使线粒体内的TDP-43水平增加、并加重TDP-43诱导的线粒体损伤和退行性表型(Wang et al., PLoS Genetics, 2019)。
TDP-43蛋白可被转运至线粒体并引起线粒体损伤,但是对于TDP-43如何进入线粒体仍然知之甚少。此外,线粒体损伤是TDP-43在线粒体错误定位引起的,还是线粒体介导的TDP-43 降解的副作用,仍有待深入研究。
2023年11月11日,中国科学院生物物理研究所朱笠研究组在《Cell Death & Disease》期刊在线发表了题为"Integration of FUNDC1-associated mitochondrial protein import and mitochondrial quality control contributes to TDP-43 degradation"的研究论文,该研究报道了线粒体自噬受体FUNDC1参与调节TDP-43进入线粒体及降解的分子机制,为进一步理解神经元细胞中TDP-43的稳态调控机制提供线索。
研究人员通过生物信息学分析、结合细胞模型和果蝇模型系统地研究了线粒体自噬受体FUNDC1调节TDP-43蛋白质稳态的分子机制。对GEO数据库中TDP-43蛋白病RNA-seq数据进行相关性分析和共表达网络构建,发现线粒体自噬受体基因FUNDC1与TDP-43以及HSP70/HSP40/TOM家族的多个成员存在共表达关系(图1)。在细胞模型中,尽管FUNDC1与TDP-43没有直接的蛋白-蛋白相互作用,但是过表达TDP-43会增加FUNDC1的水平,且FUNDC1促进TDP-43的线粒体定位。生化实验表明,FUNDC1和TDP-43可分别与TOM70、HSPA8和DNAJA2相互作用,而且后三种蛋白与TDP-43的线粒体输入密切相关。进一步实验证明,FUNDC1通过促进TDP-43-TOM70和DNAJA2-TOM70的相互作用来增加TDP-43的线粒体定位,且此效应独立于FUNDC1的LC3相互作用区域LIR。在TDP-43蛋白病转基因果蝇模型中,过表达FUNDC1会增强TDP-43诱导的线粒体损伤,而下调FUNDC1可部分挽救TDP-43诱导的线粒体损伤。在类神经元细胞SH-SY5Y中,过表达FUNDC1通过增加线粒体蛋白酶LONP1水平和激活线粒体自噬来调节细胞质中TDP-43的蛋白水平。总之,FUNDC1通过调节与线粒体蛋白输入途径相关的蛋白质机器促进TDP-43的线粒体输入,且FUNDC1介导的线粒体质量控制在TDP-43蛋白的稳态调控中发挥重要作用(图2)。
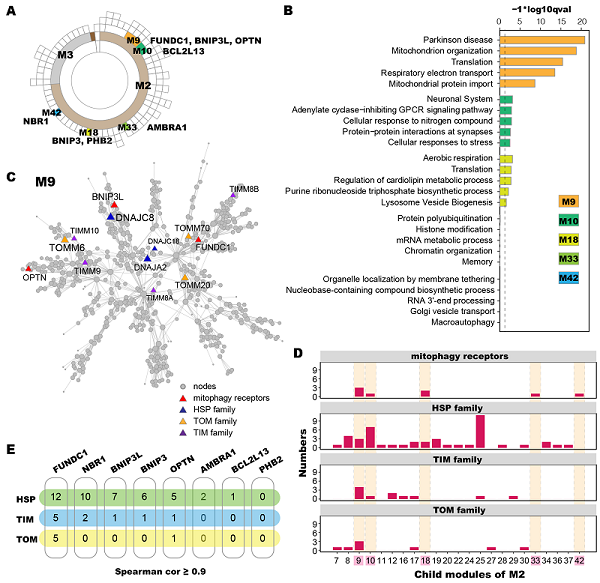
图1. TDP-43蛋白病脑样本RNA-seq数据的共表达网络分析
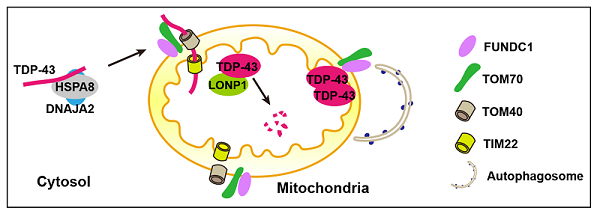
图2. FUNDC1促进TDP-43线粒体输入并调控其蛋白稳态的模式图
中国科学院生物物理研究所朱笠研究团队已毕业博士生马金法为本文的第一作者,朱笠研究员和美国西北大学医学院吴瑛教授为共同通讯作者,中国科学院动物研究所刘垒研究员和南开大学生命科学学院陈佺教授为本文做出了重要贡献,研究组成员刘江红、宋露和杨凌垚也为本文做出贡献。该研究工作得到了国家重点研发计划和国家自然科学基金项目的资助。电镜实验得到生物物理所生物成像中心的协助。
文章链接:
https://www.nature.com/articles/s41419-023-06261-6
(供稿:朱笠研究组)
附件下载: