新技术实现蛋白质在细胞内的高通量、高分辨结构解析
发布时间:2023-03-16
冷冻电镜单颗粒技术已经实现了解析溶液中蛋白质的结构达到近原子分辨率,甚至在apoferritin上达到了原子分辨率。然而,在工作状态的细胞环境中的蛋白质往往具有更多的原生构象,并可能形成比纯化蛋白质更完整的复合物,因此,实现高分辨的原位蛋白质结构解析是目前冷冻电镜的一个重要目标。
冷冻电子断层(cryo-ET)结合亚单位平均是目前实现蛋白质原位结构解析的通用方案,但是断层要求对每一个区域收集倾转序列,这严重降低了原位的数据采集通量,并且倾转序列之间存在对中误差和冰层畸变等问题,导致分辨率难以突破。
为了实现原位蛋白质的高通量、高分辨率结构解析,中国科学院生物物理所章新政组一直致力于开发不基于电子断层的新型原位结构解析算法,2023年3月15日,《Nature Communications》杂志在线发表了题为"Determining protein structures in cellular lamella at pseudo-atomic resolution by GisSPA" 的研究论文,该论文基于课题组2021年发表的isSPA算法进行了GPU加速优化,将计算效率提高了约400-500倍。
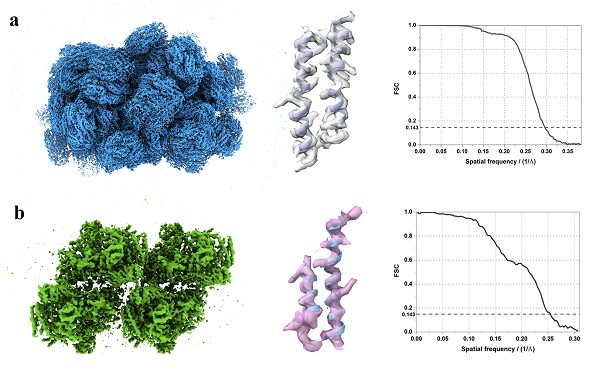
图1. 藻胆体(a)与光系统II(b)的高分辨原位结构
论文将FIB减薄的细胞样品作为测试数据,使用GisSPA分别解析了细胞中的分子量约为14.7 MD的藻胆体和分子量约为1.5MD的光系统II复合物的高分辨结构,其分辨率分别为3.4埃和3.9埃(图1)。此外,论文中还探索了GisSPA算法针对不同分子量大小的蛋白质和切片厚度的适用范围,结论认为在切片厚度为100-150纳米时,可解析的最小复合物的分子量约为1.1 MD(图2)。该方法较断层重构数据采集效率提升30~40倍,解析分辨率也获得显著提升。
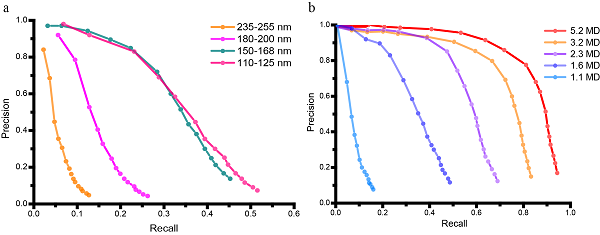
图2. GisSPA算法探测效率与切片厚度(a)和蛋白质分子量(b)的相关性
中科院生物物理研究所的章新政研究员和北京理工大学的万晓华副研究员为该论文的通讯作者,章新政组博士后程静和中科院计算所研究生刘童为共同第一作者。该工作受到国家重点研发计划、国家自然科学基金委、中国科学院战略性先导科技专项(B类)、中国科学院基础前沿科学研究计划等资助。
论文中的FIB样品来自于清华大学的隋森芳团队。隋森芳团队应用isSPA算法与电子断层的结合研究了红藻的PBS-PSII-PSI-LHC巨型复合物,其中isSPA算法将复合物的分辨率提高至3.3埃。该工作同日在《Nature》杂志在线发表,题为"In situ structure of the red algal phycobilisome-PSII-PSI-LHC megacomplex",清华大学隋森芳院士、王宏伟教授和中科院生物物理所章新政研究员为共同通讯作者。清华大学隋森芳课题组游鑫、王宏伟课题组张星、中科院生物物理所章新政组程静和南方科技大学隋森芳课题组肖亚男为共同第一作者。
章新政课题组一直致力于新型原位结构解析技术的开发,早期基于此开发了一种蛋白质识别算法,并对探测函数的最优权重进行了推导,新函数提升了叠合密度中探测目标蛋白的效率。结合排序函数减少蛋白质识别算法引入的假阳性数据,有效减轻模型偏差的影响,实现高分辨重构,该工作于2021年发表在《The Innovation》杂志(https://doi.org/10.1016/j.xinn.2021.100166)。更广泛的原理介绍相关论文发表在《Biophysics Reports》上(https://doi.org/10.52601/bpr.2021.210001)。
文章链接:
Determining protein structures in cellular lamella at pseudo-atomic resolution by GisSPA
https://doi.org/10.1038/s41467-023-36175-y
In situ structure of the red algal phycobilisome-PSII-PSI-LHC megacomplex
https://doi.org/10.1038/s41586-023-05831-0
(供稿:章新政研究组)
附件下载: