张宏课题组发现内质网塑形蛋白ATL2/3调控自噬起始位点的形成
发布时间:2021-05-17
2021年5月14日,《Journal of Cell Biology》杂志在线发表了中国科学院生物物理研究所张宏课题组题为"Atlastin 2/3 regulate ER targeting of the ULK1 complex to initiate autophagy"的研究论文,该文揭示了ATL2/3与参与自噬起始的ULK1复合体直接相互作用,促进其在内质网上组装,从而介导自噬体的形成。
自噬(autophagy)是真核细胞中由溶酶体介导的降解途径。自噬通过形成双层膜的自噬体以包裹部分的细胞质组分,比如受损的细胞器、错误折叠的蛋白等物质,并将其运送到溶酶体进行降解。内质网在多细胞生物的自噬过程中发挥重要的功能:当细胞感受到应激刺激等自噬诱导信号时,ULK1复合体首先被招募到内质网上,形成自噬起始位点,进而招募下游的自噬蛋白共同诱导隔离膜的形成;在隔离膜的延伸过程中,ULK1复合体与WIPI2蛋白相互作用,介导隔离膜与内质网的膜接触的形成,为自噬体的形成提供脂来源。哺乳动物细胞中,ULK1复合体主要包括FIP200、ULK1、ATG13和ATG101蛋白,这四个蛋白相互作用,动态地结合在一起。目前ULK1复合体在内质网上动态组装的机制尚不清楚。
内质网塑形蛋白Atlastin是一类dynamin超家族蛋白,该家族由ATL1、ATL2和ATL3组成,负责调控内质网的融合,ATLs的缺失会造成内质网形态的改变。课题组发现敲除ATL2/3会抑制自噬活性。自噬诱导后,细胞质中弥散分布的LC3I经过脂化作用,以LC3II的形式偶联到自噬体膜上。研究发现,在ATL2/3双敲细胞中,LC3II/LC3I比例明显降低,脂化过程减慢。同时,自噬底物p62明显累积,自噬体数量减少,表明敲除ATL2/3抑制自噬的活性。随后研究发现,与对照细胞相比,在ATL2/3双敲细胞中,FIP200和ATG13的内质网招募没有明显改变,但ULK1与ATG101的招募显著减少。ATG13-ULK1及ATG13-ATG101之间的相互作用也明显减弱。进一步研究表明ATL2/3帮助ATG13招募ULK1与ATG101。该研究还发现在ATL2/3双敲细胞中,WIPI2和ULK1及FIP200之间的相互作用减弱,电镜结果表明自噬体/隔离膜与内质网膜接触也减少,说明敲除ATL2/3抑制内质网与隔离膜接触的形成。
这项研究揭示了内质网蛋白ATL2/3调控自噬的分子机制。ATL2/3与ATG13和ULK1相互作用,促进ULK1复合体在内质网的招募,参与内质网隔离膜接触位点的形成。该工作揭示了自噬体形成过程中,ULK1复合体在内质网上的动态组装的分子机制。
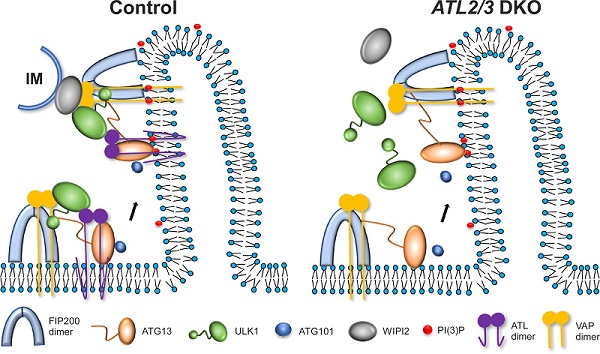
图 ATL2/3参与自噬调控的工作模型
中国科学院生物物理研究所张宏研究员和胡俊杰研究员为本文的共同通讯作者,生物物理所博士生刘楠为本文的第一作者。该项目受到国家自然科学基金、科技部、北京市科委、中科院先导及中科院前沿局等的支持。
文章链接:https://doi.org/10.1083/jcb.202012091
(供稿:张宏研究组)
附件下载: