 最新图片新闻
最新图片新闻去甲肾上腺素(Noradrenaline,NA)是神经系统中重要的单胺类神经递质。在中枢神经系统中,去甲肾上腺素能神经元起始于脑干中一个名为"蓝斑核"的细胞核团,并广泛投射至大脑的其他区域,调节情绪、注意力、记忆、性欲等多种神经活动。此外,NA在背根神经节能够与伤害传入神经元上的α2A受体结合,抑制疼痛信号的传递。在去甲肾上腺素能神经元中,NA由突触前膜释放到突触间隙,激活去甲肾上腺素受体,进而启动一系列信号传导过程。定位于突触前膜上的去甲肾上腺素转运体(NET),通过利用细胞内外存在的钠离子和氯离子的浓度梯度,可以将NA重新摄取回突触前神经元内,进而终止神经信号的传递。NET对于NA的回收维持了神经系统中NA水平的平衡,使得神经系统能够正常行使生理功能。目前,临床上有多种小分子药物通过抑制NET 来提高突触间隙 NA 的浓度,进而增强去甲肾上腺素能神经元的信号传递,从而治疗抑郁症、注意力缺陷多动障碍等疾病。此外,多肽类毒素χ-MrA能以非竞争方式特异地抑制 NET,通过提高伤害传入神经元的 α2A 受体活性,阻断疼痛信号的传递,展现出显著的镇痛效果。然而,目前对于 NET转运底物以及与不同药物分子结合模式仍不清楚。

图1:去甲肾上腺素能神经元
2024年7月31日,中国科学院生物物理研究所赵岩团队与丹麦哥本哈根大学Claus Loland团队合作在《Nature》发表一篇题为"Transport and inhibition mechanisms of the human noradrenaline transporter"的研究论文。研究人员通过单颗粒冷冻电镜技术解析了人源去甲肾上腺素转运体(NET)在未结合底物(NETapo),结合底物去甲肾上腺素(NETNA)、结合芋螺毒素χ-MrlA类似物(NETMrlA)、结合安非他酮(NETBPP),以及结合齐拉西酮(NETZPD)的复合物高分辨率结构。这些复合物结构揭示了NET在多种功能状态的结构特征以及不同药物分子的结合模式,深入地阐释了NET的转运机制,并为新型药物的开发提供了结构基础。
研究人员解析了内向开口状态的NETapo及NETNA结构,其分辨率均为2.6 Å。与底物的复合物结构揭示了NET在中心结合位点识别底物NA的机制。与此同时,研究人员还意外地发现在胞外侧存在另外一个构象特异的底物结合位点,该位点只存在于内向开口状态。在外向开口状态,由于局部构象变化,该底物结合位点消失。将该位点相互作用的关键氨基酸突变后,NET的转运活性降低,表明该位点可能对转运功能具有调节作用。此外,NET是依赖于钠离子、氯离子的次级转运蛋白。研究人员解析了NET在不同转运功能状态下离子的结合位点,这为理解离子、底物的结合与构象变化的偶联机制提供了结构基础。
χ-MrlA是从海洋软体动物芋螺中提取的一种由13个氨基酸组成的多肽类毒素,它是SLC6家族中唯一已知的多肽类抑制剂,也是NET唯一已知的非竞争性抑制剂。基于χ-MrlA,Xenome公司开发的类似物XEN2174可用于术后疼痛、癌症疼痛和慢性疼痛的治疗。通过解析与χ-MrlA的复合物结构,研究人员发现 χ-MrlA类似物将NET稳定在外向开口的构象,深入结合在NET胞外的口袋中,和NET形成了广泛的相互作用。与底物结合的复合物结构对比发现,χ-MrlA的结合位置与底物分子并没有重合,这也从结构水平解释了χ-MrlA是NET的非竞争性抑制剂。结合结构分析以及突变体功能实验验证,研究人员还揭示了χ-MrlA选择性抑制NET,但不抑制同家族转运蛋白血清素转运体(Serotonin transporter,SERT)和多巴胺转运体(Dopamine transporter,DAT)的关键结构基础。这些信息为靶向单胺类转运体家族的新型多肽类药物的研究提供了重要的结构基础。
临床上常用的抗抑郁药物包括三环类抑制剂、选择性血清素再摄取抑制剂(SSRI)及血清素-去甲肾上腺素再摄取抑制剂(SNRI)。这些药物主要通过抑制SERT达到治疗抑郁症的效果,但是有时导致一些副作用,如恶心、头痛、失眠和口干等。安非他酮是临床上唯一选择性抑制NET和DAT的抗抑郁药,可以避免抑制SERT带来的副作用。此前的研究发现,抗精神病药物齐拉西酮能够同时抑制NET、DAT和SERT。为了揭示两种药物分子选择性抑制三种单胺类神经递质转运蛋白的分子机制,研究人员解析了NET与安非他酮及齐拉西酮复合物结合。结构分析发现,与经典的抗抑郁药物不同,安非他酮和齐拉西酮都结合在NET朝向胞内侧开口的结合口袋中。通过比较两种药物分子的结合模式,研究人员发现安非他酮的正辛基团和齐拉西酮的乙基都位于TM6和TM8之间的卡口中。相比之下,安非他酮的正辛基团体积较齐拉西酮的乙基更大。在SERT中,由于卡口一侧的氨基酸从甘氨酸变为丙氨酸,该卡口变得更加狭窄,导致安非他酮无法有效抑制SERT,但这一变化并未影响齐拉西酮的抑制效果。这些发现为靶向MATs药物开发提供了关键信息。
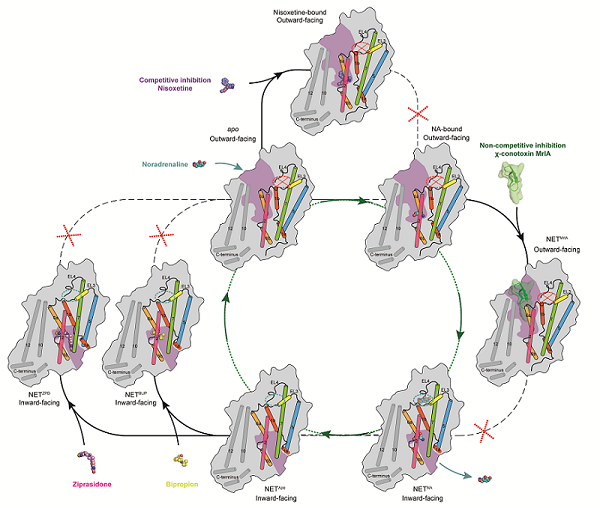
图2:NET的转运循环及多种抑制模式
中国科学院生物物理研究所赵岩研究员以及哥本哈根大学的Claus Loland教授为该论文的共同通讯作者,中国科学院生物物理研究所博士研究生胡拓、于卓亚和北京大学现代农业研究院赵珺研究员为论文的共同第一作者。此外,生物物理所的博士生孟宇飞、魏一青、白秦儒,丹麦哥本哈根大学的Kristine Salomon,中国海洋大学的于日磊教授、博士研究生张景会,军事医学科学院的戴秋云研究员、徐书静同学,以及首都医学科学创新中心的杨蓓老师也为该研究提供了帮助。该研究得到科技创新2030"脑科学与类脑研究"重大项目、国家重点研发计划项目、中国科学院战略性先导科技专项(B类)、国家自然科学基金项目的资助。冷冻电镜数据收集得到中国科学院生物物理研究所蛋白质科学研究平台生物成像中心、北京大学现代农业研究院生物微观结构研究平台的技术支持。放射性转运实验得到中国科学院生物物理研究所放射性同位素实验室的帮助。
文章链接:https://www.nature.com/articles/s41586-024-07638-z
(供稿:赵岩研究组)
 附件下载:
附件下载: