 科普报道
科普报道阿尔茨海默病(Alzheimer's disease,AD)是一种以进行性认知功能障碍和记忆减退为主要特征的神经退行性疾病,已成为威胁中老年人身心健康的主要疾病之一。AD以细胞外β淀粉样蛋白(amyloid β,Aβ)异常沉积形成老年斑、细胞内Tau蛋白过度磷酸化形成神经原纤维缠结(neurofibrillary tangle,NFT)及神经元丢失等为主要的病理特征,临床表现为记忆障碍、失语、失用、失认、视空间技能损害、执行功能障碍以及人格和行为改变等。目前,我国每年平均有30万新发AD病例。随着人口老龄化日益严重,AD已成为全球最重要的社会医学问题之一。AD是一种由遗传因素(载脂蛋白E4、淀粉样前体蛋白、早老素1、早老素2等基因突变)和非遗传因素(年龄、吸烟、嗜酒等因素刺激)相互作用引起的复杂疾病,但其发病机制尚不清楚,也尚无治疗AD的有效手段。携带相似或相同易感基因的个体在发生AD风险、其病理程度和临床表现中出现差异,提示表观遗传修饰可能在AD的异质性中发挥作用。

什么是表观遗传修饰
表观遗传修饰是指在不改变DNA序列的情况下,受环境因素影响导致基因表达发生可遗传改变。表观遗传修饰主要包括DNA甲基化、组蛋白甲基化、组蛋白乙酰化、RNA修饰和非编码RNA等。从DNA到组蛋白,再到RNA,这些发生在不同维度的化学修饰过程相互作用,最终影响突触、记忆、神经通路以及神经信号相关蛋白质的改变。大量研究表明,表观遗传学在AD发生发展中发挥重要作用【1】。

DNA修饰与AD
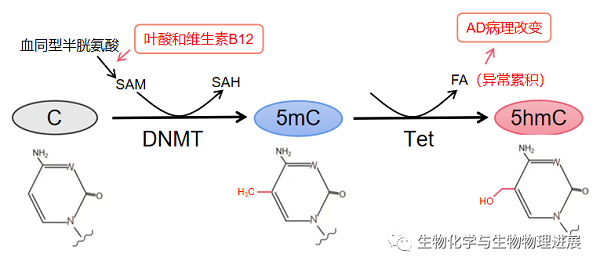
DNA甲基化和去甲基化在神经发育和维持大脑突触可塑性过程中起重要作用。DNA甲基化是指在DNA甲基转移酶(DNA methyltransferase,DNMT)的作用下,由S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)提供一个甲基基团,与DNA序列上特定的碱基共价结合的化学修饰过程。DNA甲基化生成物质主要包括5mC、N4-甲基胞嘧啶(N4-methylcytosine,4mC)和N6-甲基腺嘌呤(N6-methyladenine,6mA)。在哺乳动物中,目前研究比较多的是发生于胞嘧啶(C)-磷酸(p)-鸟嘌呤(G)二核苷酸(CpG)上胞嘧啶环第5号碳原子上甲基化修饰(5mC)。DNA去甲基化主要通过Tet蛋白家族催化5mC转变为5hmC,5hmC还可以进一步氧化形成5甲酰基胞嘧啶(5-formylcytosine,5fC)和5羧基胞嘧啶(5-carboxylcytosine,5caC),进而产生未甲基化的胞嘧啶C。因此,5hmC也是一种DNA去甲基化的中间代谢产物。5hmC在正常神经发育和衰老过程中保持动态平衡,且其修饰水平的改变在一定程度上与AD病理变化相关。SAH、PSEN1和BACE等基因的DNA低甲基化修饰导致其表达水平升高,从而导致Aβ沉积。而Aβ沉积使与Aβ降解相关的基因如NEP发生高甲基化,进一步加重Aβ沉积。同时,DNA去甲基化过程中伴随活性物质甲醛(formaldehyde,FA)生成,若FA异常累积,将引起Aβ沉积、Tau蛋白磷酸化,从而诱发AD【2】。同时,Tet蛋白家族通过调控不同类型细胞中DNA的去甲基化,如Tet1调控神经元和NSCs、Tet2调控aNSCs、Tet3调控NPCs等而影响神经再生。因此,大脑中5mC和5hmC水平变化影响Aβ生成、Tau蛋白磷酸化和神经再生,引起AD病理改变。
适当补充叶酸和维生素B12可减少AD发病的机率。若叶酸和维生素B12严重缺乏,可降低Aβ生成相关的β-分泌酶1(beta-site APP cleaving enzyme 1,BACE1)和γ-分泌酶的甲基化水平,从而促使Aβ沉积【3】。而Aβ沉积使Aβ降解酶如中性内肽酶(neprilysin,NEP)高甲基化,并下调NEP表达,进一步加重Aβ沉积。因此,提高机体叶酸、维生素B12含量,有利于减少Aβ斑块生成,对抗AD认知功能障碍。
组蛋白修饰与AD
组蛋白是一种由H2A、H2B、H3和H4组成的八聚体,可与DNA交联形成核小体。组蛋白在其N端尾部发生修饰,影响染色体三维结构,最终导致基因转录改变。目前研究最广泛、与AD关联最密切的是组蛋白甲基化/去甲基化以及组蛋白乙酰化/去乙酰化过程。
组蛋白甲基化和去甲基化是指在组蛋白甲基化转移酶(histone methyltransferase,HMT)和组蛋白去甲基化酶(histone demethylase,HDM)作用下分别添加和脱去甲基基团的过程。在AD患者大脑内,组蛋白甲基化/去甲基化与认知记忆能力紧密相关,通过作用于G9a/GLP复合体、SET1/MLL HMT家族以及LSD1、JMJD3,分别影响认知相关基因表达以及神经元增殖分化,从而进一步改善AD认知障碍【4-7】。
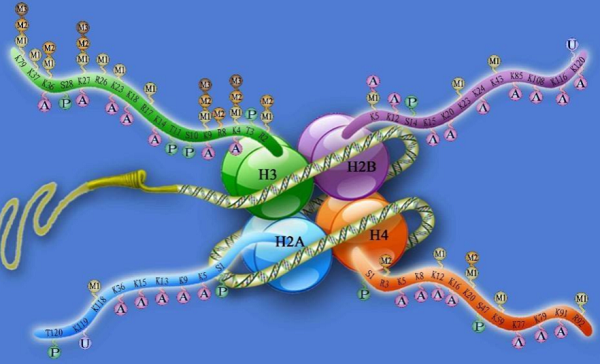
另外,组蛋白乙酰化/去乙酰化在调节AD患者神经病理和记忆能力发面发挥重要作用。组蛋白乙酰化/去乙酰化修饰过程分别由组蛋白乙酰转移酶(histone acetylases,HATs)和组蛋白去乙酰酶(histone deacetylases,HDACs)催化产生。利用药物提高HATs的活性,有助于短期和长期记忆的形成。HDAC2、HDAC3和HDAC6是记忆巩固的负调节剂。利用HDACis抑制HDAC2、HDAC3和HDAC6表达,可以清除Aβ和Tau蛋白,有效对抗神经病理和认知障碍【8】。
RNA修饰与AD
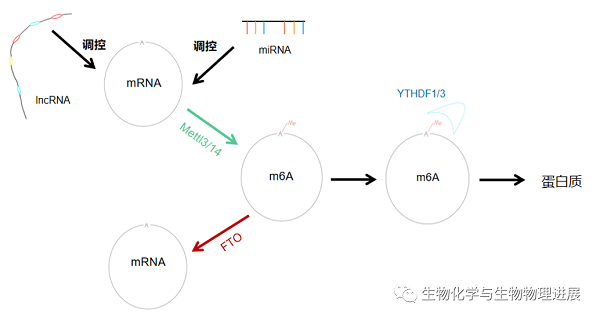
发生在mRNA上的N6-甲基腺苷(N6-methyladenosine,m6A)修饰能否诱发AD?这是近年来科学家一直在探求的问题。尽管m6A在原核生物中的研究已经十分透彻,但其对真核生物功能影响的探索才刚刚起步。
实际上,m6A修饰过程能够直接或间接地影响神经系统发展和演化,包括神经分化功能以及突触可塑性。m6A甲基转移酶Mettl3、Mettl14和去甲基化酶FTO协同作用影响m6A修饰水平,间接调节神经元分化及突触可塑性,从而影响认知能力。m6A结合蛋白YTHDF1和YTHDF3则在翻译水平上直接影响突触蛋白合成,从而改变神经系统信息传递效率,调控记忆发生。
另外,在AD患者外周循环(血清、血浆、外泌体、全血、外周血单核细胞)和脑脊液中能够检测到异常表达的非编码RNA(non-coding RNA,ncRNA)。ncRNA是指无法翻译蛋白质的RNA分子,其中microRNA(miRNA)和长链非编码RNA(long non-coding RNA,lncRNA)被证实是AD的理想生物标志物。异常表达的ncRNA引起Aβ聚集、Tau磷酸化、神经炎症、突触可塑性和自噬等病理生理过程。血浆BACE1-AS含量改变可诊断AD进展情况,同时BACE1-AS上调与Aβ生成形成正反馈机制,加速机体细胞损伤和凋亡机制【9】;脑组织过表达BC200,提高BACE1表达以加速Aβ沉积【10】。lncRNA BDNF-AS和GDNFOS则分别通过抑制BDNF和GDNF表达,抑制神经发育【11-12】。microRNA(miRNA)是长度为22个核苷酸的小分子ncRNAs,广泛存在于血液、外泌体、脑脊液(CSF)、脑组织等部位。目前已被证明是一种参与AD发生的潜在调节因子。大部分miRNA(miR-29、miR-31、miR-101、miR-346等)直接或通过调控BACE1间接影响Aβ沉积,部分miRNA(miR-34a、miR-219、miR-128a、miR-125b、miR-124)间接调控Tau表达或直接影响Tau蛋白过度磷酸化,miR-137调控突触可塑性以及miR-142影响炎症发生,从而在多方面引起AD病理改变。
展望
表观遗传修饰是一种动态、可逆的变化过程,因此通过干预逆转异常的表观修饰,可达到改善AD的目的。临床分别采集AD患者CSF、外周血或脑组织样本进行检测,发现异常的DNA修饰、组蛋白修饰、RNA修饰、ncRNAs等表观遗传修饰与AD发生发展密切相关,揭示了表观遗传修饰的改变与AD风险呈显著的相关性且可用于预测或诊断AD。应用药物、物理刺激或遗传学等手段干预调控各种修饰酶的表达水平,引起全基因组表观遗传修饰的广泛改变,但不能精确调控单基因的表观修饰,因此,目前尚缺乏特异性操控单基因表观遗传修饰的干预手段。研发以单基因、单一组蛋白或单一lncRNA或miRNA的表观遗传修饰为靶标的干预手段,更有助于理解表观遗传机制的具体作用过程。此外,在表观遗传机制中DNA修饰、组蛋白修饰、RNA修饰和ncRNA之间复杂的相互作用共同调控下游靶基因的表达,因此,未来研究需全面而系统地探究不同表观修饰在AD发生发展进程中内在关系。因此,未来研究热点将投射到研发有效的、安全的、低成本的操纵全基因组表观修饰的药物,或研发特异性操纵单个表观遗传学靶标的干预方法,真正实现干预可行性、特异性和临床有效性三者合一的治疗效果。(详情请点击阅读原文)
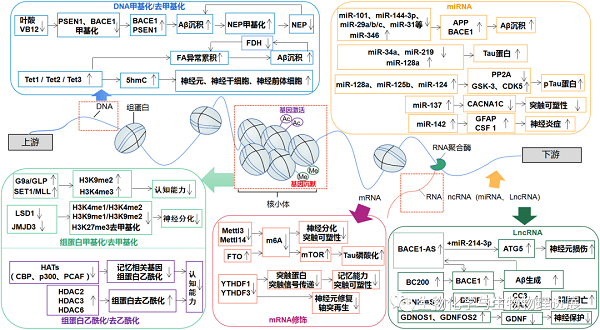
表观遗传修饰在AD中的作用
参考文献
[1] Mayo S, Benito-León J, Pe?a-Bautista C, et al. Recent evidence in epigenomics and proteomics biomarkers for early and minimally invasive diagnosis of Alzheimer's and Parkinson's diseases. Curr Neuropharmacol, 2021, 19(8): 1273-1303
[2] Tong Z, Han C, Qiang M, et al. Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer's disease. Neurobiol Aging, 2015, 36(1): 100-110
[3] Fuso A, Seminara L, Cavallaro R A, et al. S-adenosylmethionine/ homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci, 2005, 28(1): 195-204
[4] Sharma M, Dierkes T, Sajikumar S. Epigenetic regulation by G9a/GLP complex ameliorates amyloid-beta 1-42 induced deficits in long-term plasticity and synaptic tagging/capture in hippocampal pyramidal neurons. Aging Cell, 2017, 16(5): 1062-1072
[5] Cao Q, Wang W, Williams J B, et al. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer's disease. Sci Adv, 2020, 6(50): eabc8096
[6] Longaretti A, Forastieri C, Toffolo E, et al. LSD1 is an environmental stress-sensitive negative modulator of the glutamatergic synapse. Neurobiol Stress, 2020, 13: 100280
[7] Park D H, Hong S J, Salinas R D, et al. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep, 2014, 8(5): 1290-1299
[8] De Simone A, Milelli A. Histone deacetylase inhibitors as multitarget ligands: new players in Alzheimer's disease drug discovery?. ChemMedChem, 2019, 14(11): 1067-1073
[9] Li F, Wang Y, Yang H, et al. The effect of BACE1-AS on β-amyloid generation by regulating BACE1 mRNA expression. BMC Mol Biol, 2019, 20(1): 23
[10] Li H, Zheng L, Jiang A, et al. Identification of the biological affection of long noncoding RNA BC200 in Alzheimer's disease. Neuroreport, 2018, 29(13): 1061-1067
[11] Modarresi F, Faghihi M A, Lopez-Toledano M A, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol, 2012, 30(5): 453-459
[12] Airavaara M, Pletnikova O, Doyle M E, et al. Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J Biol Chem, 2011, 286(52): 45093-45102
作者简介
林苏扬:宁波大学医学院临床医学专业本科生。现阶段在宁波大学医学院浙江省病理生理学技术研究重点实验室,主要致力于表观遗传修饰影响阿尔茨海默病的相关研究。
李丽萍:博士,硕士生导师,宁波大学医学院生理与药理学科教师。主要从分子、细胞及动物模型水平研究在生理或病理条件下表观遗传修饰对阿尔茨海默病的神经病理、突触可塑性和认知功能调控作用的研究。
(作者:林苏扬、李丽萍)
 附件下载:
附件下载: