刘光慧研究组利用干细胞技术
发布时间:2014-07-10
2014年7月7日,中国科学院生物物理研究所刘光慧研究组及其合作者在Nature Communication杂志发表了关于范可尼贫血症(Fanconi Anemia,FA)转化医学研究的重要成果,首次提出多组织干细胞加速衰老或衰竭是FA的根本性病因,并基于此发展出相应的干细胞、基因和药物治疗策略。
FA属先天再生障碍性贫血,为严重的常染色体隐性遗传病,是由编码FA通路系列DNA修复蛋白的基因发生突变所致。临床表现为骨髓造血功能衰竭、多发性先天畸形及肿瘤易感性增加等。目前临床上尚无针对FA的有效治疗药物。
体细胞重编程和基因组靶向编辑技术的结合为治疗遗传性血液疾病带来了希望。但对FA而言,该治疗策略一直以来面临着难以克服的技术瓶颈。首先,FA通路的缺陷构成了体细胞重编程的严重障碍,科学家一度认为FA患者的体细胞是无法被直接重编程的。人们迄今尚无法衍生出非基因组整合型(integration-free)的FA诱导性多能干细胞(iPSC)。其次,FA的体细胞缺乏有效的DNA同源重组机制,故而增加了原位矫正其致病基因突变的难度。
在该项研究中,刘光慧团队从克服体细胞重编程所伴随的细胞衰老入手,首次利用非基因组整合技术实现了FA成纤维细胞的表观遗传重编程。同时,研究人员还突破了DNA同源重组效率低下的技术障碍,在FA-iPSC中成功实现了对致病基因FANCA的靶向矫正。安全性评价结果显示,整个重编程和基因矫正过程最大限度地保持了患者基因组的完整性。研究还揭示了FA患者发生严重再生障碍性贫血的根源,即不仅归因于造血干细胞的过早衰竭,还与骨髓造血微环境间充质干细胞的加速衰老密切相关。此外,间充质干细胞的缺陷有效解释了FA所伴随的多发性先天畸形(如骨发育畸形等)。最后,携带FANCA突变的神经干细胞表现为向神经元定向分化能力的减弱及肿瘤相关基因的激活,从而为FA相关的头小畸形、智力发育迟缓和脑部肿瘤等表型提供了理论依据。
该研究不仅在人类组织干细胞水平揭示了FA的新型病因学基础,而且为FA的干预和治疗提供了全新的研究平台和解决方案。结果显示,靶向矫正FANCA基因突变可以从根本上逆转FA的病理表型,使干细胞的DNA修复能力、基因表达谱、“干性”及其它功能得以恢复。这样,经遗传修复的造血干细胞可能在将来被应用于自体移植,以治疗FA病人的骨髓衰竭和再障性贫血(疾病最大死因)。更为重要的是,该研究利用人类疾病模型成功地筛选得到了可抑制FA干细胞加速衰老或衰竭的新型小分子化合物。药理学实验显示,这些化合物具有抑制FA细胞表达炎性因子和促凋亡因子的活性。由于iPSC向造血干细胞的定向分化及后续的细胞移植的方法尚不成熟,因此基于FA人类疾病模型所筛选获得的候选化合物(药物)可能更有希望在短期内应用于临床实验和治疗。
迄今为止,刘光慧团队已成功建立儿童早衰症、核纤层疾病、贫血症和帕金森氏症等人类疾病的干细胞研究模型和药物评价体系。这一最新成果将为建立新型的FA的基础和转化医学研究模式奠定基础。
刘光慧研究员和美国Salk研究所的Juan Carlos Izpisua Belmonte教授为论文的共同通讯作者。研究得到科技部、基金委及中科院干细胞与再生医学战略先导专项等资助,相关研究成果已申请国家发明专利。
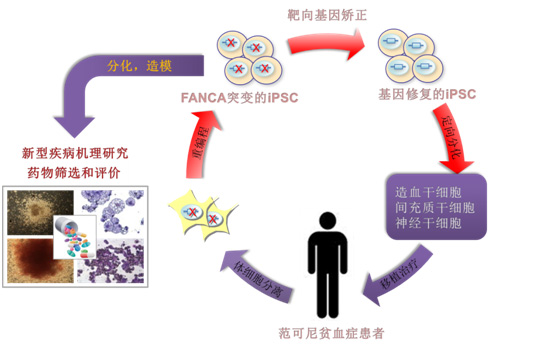
图:基于干细胞技术的范可尼贫血症的机理研究和治疗策略
文章链接:Modeling Fanconi Anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs.Nat Commun. 2014. DOI: 10.1038/ncomms5330 (http://www.nature.com/ncomms/2014/140707/ncomms5330/full/ncomms5330.html)
(供稿:刘光慧课题组)
附件下载:
下一篇: